Cystic fibrosis (CF) is a genetic disorder that affects mostly the lungs, but also the pancreas, liver, kidneys, and intestine. Long-term issues include difficulty breathing and coughing up mucus as a result of frequent lung infections.

- Genetic Disorder which is usually passed on from parents, whom maybe carriers without presenting symptoms themselves.
- Multisystem disease which affects the lungs, liver, pancreases, sweat glands and reproductive system
- Inherited disease that primarily affects the lungs and the digestive system.
Incidence
- Ireland has the highest incidence of Cystic Fibrosis in the world
- Almost 7 in every 10,000 people have the disease.
- The incidence of Cystic Fibrosis in Ireland is almost 3 times the average rate in other EU countries and the United States
- Approximately 1 in 19 Irish people are said to ‘carry’ one copy of the altered gene that causes Cystic Fibrosis
- I in 29 Caucasian Americans are carriers of CF gene
- >1158 people registered with CF in Ireland 2014.
What causes Cystic Fibrosis?
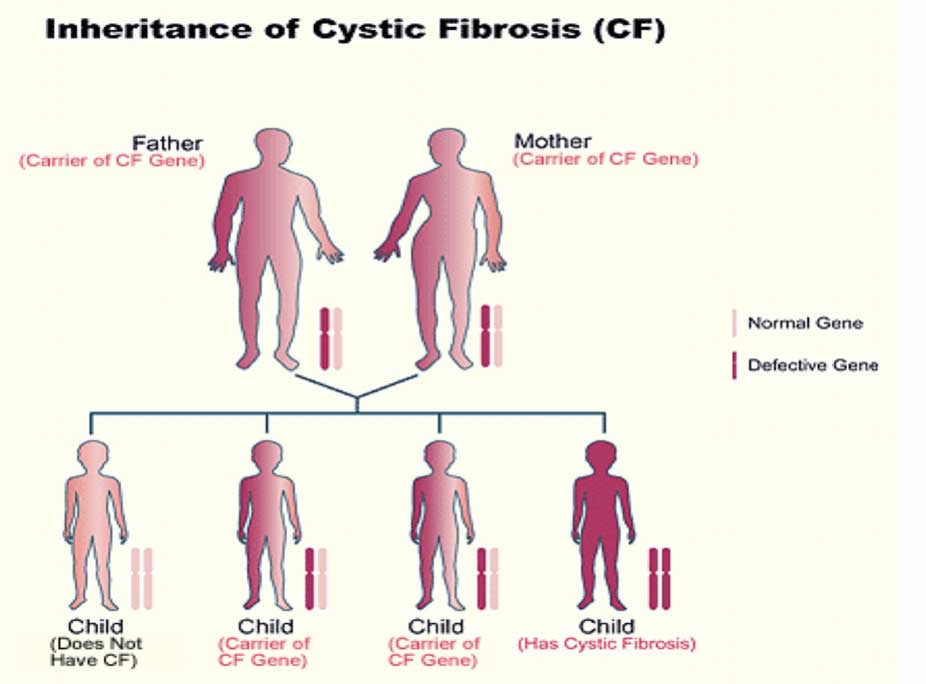
Cystic fibrosis is a genetic disease. It is inherited in an autosomal recessive manner. People with CF have inherited two copies of the defective (recessive) CF gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein.– one copy from each parent. Both parents must have at least one copy of the defective gene.
People with only one copy of the defective CF gene are called carriers, but they do not have the disease. Each time two CF carriers have a child, the chances are:
- 25 percent (1 in 4) the child will have CF
- 50 percent (1 in 2) the child will be a carrier but will not have CF
- 25 percent (1 in 4) the child will not be a carrier and will not have CF
CFTR is involved in production of sweat, digestive fluids, and mucus. When the CFTR is not functional, secretions which are usually thin instead become thick.
The defective CF gene contains a slight abnormality called a mutation. There are currently 2009 mutations identified in CF.
What are the symptoms of Cystic Fibrosis?
Cystic fibrosis signs and symptoms vary, depending on the severity of the disease. Even in the same person, symptoms may worsen or improve as time passes. Some people may not experience symptoms until adolescence or adulthood.
Respiratory signs and symptoms
The thick and sticky mucus associated with cystic fibrosis clogs the tubes that carry air in and out of your lungs. This can cause signs and symptoms such as:

- A persistent cough that produces thick mucus (sputum)
- Wheezing
- Breathlessness
- Exercise intolerance
- Repeated lung infections
- Inflamed nasal passages or a stuffy nose
- Bulbous swelling of finger tips, called clubbing.
- Cyanosis, lips and nail beds turning blue with serious hypoxia.
Respiratory system complications
- Damaged airways (bronchiectasis). Cystic fibrosis is one of the leading causes of bronchiectasis, a condition that damages the airways. This makes it harder to move air in and out of the lungs and clear mucus from the airways (bronchial tubes).
- Chronic infections. Thick mucus in the lungs and sinuses provides an ideal breeding ground for bacteria and fungi. People with cystic fibrosis may often have sinus infections, bronchitis or pneumonia.
- Growths in the nose (nasal polyps). Because the lining inside the nose is inflamed and swollen, it can develop soft, fleshy growths (polyps).
- Coughing up blood (hemoptysis). Over time, cystic fibrosis can cause thinning of the airway walls. As a result, teenagers and adults with cystic fibrosis may cough up blood.
- This condition, in which air collects in the space that separates the lungs from the chest wall, also is more common in older people with cystic fibrosis. Pneumothorax can cause chest pain and breathlessness.
- Respiratory failure.Over time, cystic fibrosis can damage lung tissue so badly that it no longer works. Lung function usually worsens gradually, and it eventually can become life-threatening.
- Acute exacerbations.People with cystic fibrosis may experience worsening of their respiratory symptoms, such as coughing and shortness of breath, for several days to weeks. This is called an acute exacerbation and requires treatment in the hospital.
Other Complications
As cystic fibrosis is a complex multi-system disease, it has many other complications including:
- Nutritional deficiency/GI abnormality
- Pancreatic insufficiency
- Obstructive Azoospermia (Male infertility)
- Cystic Fibrosis Related Diabetes
- Electrolyte abnormality (signs and symptoms include increased heart rate, fatigue, weakness and low blood pressure)
- Impaired growth/failure to thrive
- Chronic sinusitis +/- nasal polyps
- Psychological issues
Musculoskeletal Complications
With CF life expectancy improving, the prevalence of musculoskeletal complications are increasing with age. They may begin in early childhood and may affect function, quality of life and longevity.
These complications include:
- CF Bone Disease – low bone mineral density in CF secondary to Vitamin D (or K) and calcium insufficiency, malnutrition and poor growth, pancreatic insufficiency, diabetes, liver dysfunction and increased metabolic requirements due to chronic lung infection, reduced lung function and weight bearing physical activity.
- Postural Mal-alignment – altered breathing mechanics inhibit optimal postural control
- Kyphosis– prevalence increases with age/disease severity
- Back Pain – 15-70% prevalence in CF
- Increased Risk of Fractures – fracture risk factors increase with disease severity, age, decreased physical activity and aerobic capacity. Spine fractures are 100 times higher in CF than age matched general population
- Urinary incontinence – commonly reported during coughing, sneezing, exercise, lifting, laughing. Repeated coughing causes downward pressure on the pelvic floor.
Management of Cystic Fibrosis
Cystic Fibrosis is best managed by a multidisciplinary approach. This usually takes place in a specialized centre with a CF team including clinical nurse specialists, dieticians, physiotherapists, pharmacists, psychologists and social workers.
Physiotherapy in Cystic Fibrosis
Physiotherapists are vital members of the multidisciplinary team. Daily lifelong physiotherapy is effective and essential in enabling people with cystic fibrosis achieve the best quality of life; decreasing complications, reducing hospital admissions and the need for antibiotic therapy and improving exercise tolerance.
What would physiotherapy treatment for Cystic Fibrosis involve?
Here at Respiratory physiotherapy Ireland, we provide comprehensive management of people with CF from infants through to childhood. Our physiotherapists devise individualised treatment, education and training on self management and assessment and advice for musculoskeletal complications.

These may include the following:
- Airway Clearance Techniques
- Active Cycle of Breathing
- Positive Expiratory Pressure (PEP)
- Oscillating (PEP)
- Autogenic drainage
- Postural Drainage
- Manual Therapy such as percussion and vibrations
- Exercise
- Inhalation Therapy
- Advice and education on correct nebuliser and/or inhaler regime
- Inhaler technique
- Exercise Assessment and Training
- Exercise is recommended for people with CF throughout their lifespan and has a positive effect on pulmonary function
- Musculoskeletal Complications
- Assessment and advice on secondary complications such as bone health, urinary incontinence, back pain and postural impairments
- Physiotherapy Management of Continence
- Non-Invasive Ventilation
- Lung Transplantation
- End of Life Care
- May include home visits if unable to attend specialised CF centre
Summary
At Respiratory Physiotherapy Ireland, our respiratory physiotherapists can provide specialist assessment and treatments for people with cystic fibrosis. Physiotherapy is a key member of the multidisciplinary team (MDT)in managing CF. All treatments provided here are as part of and in addition to the patient’s own CF MDT management in CF centres.